INSERT-seq
help page
INSERT-seq is a bioinformatics pipeline that can be used to analyse the landscape of genomic integrations from samples sequenced following INSERT-seq library prep protocol.
A command line version of the pipeline is available to download in a Bitbucket repository.
Website usage
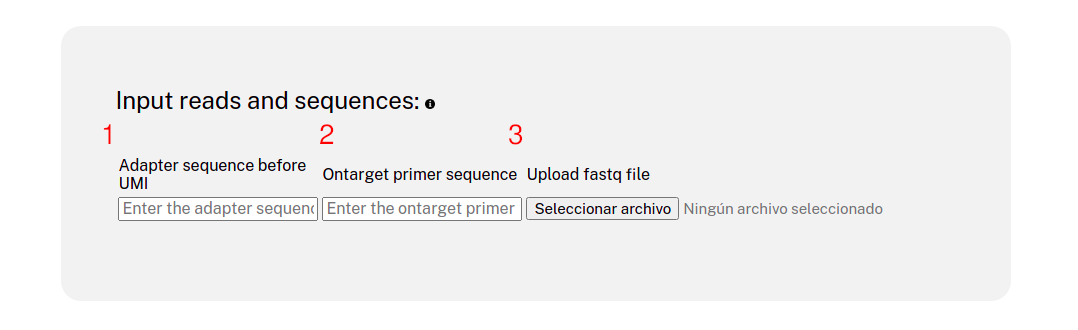
- 1. Adapter sequence before UMI: Write the sequence of the adapter part before the UMI.
- 2. Ontarget primer sequence: Write the on-target primer to detect the integrated sequence and trim it.
- 3. Upload fastq file: Upload one file per sample. The file name must have the form SampleID.fastq. The file must contain basecalled Oxford Nanopore reads obtained by following the INSERT-seq library-prep protocol. For each sample, provide the adapter sequence and the ontarget sequence in the previous fields.
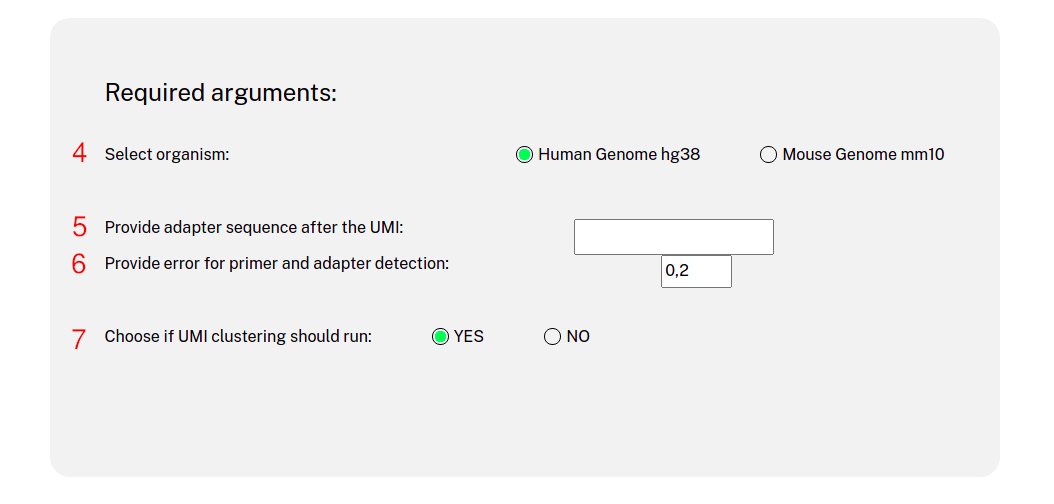
- 4. Select the reference organism. Currently only humand and mouse genomes are available.
- 5. Provide the second adapter sequence, the part of the adapter after the UMI.
- 6. You can change the error rate for primer and adapter detection, by default 0.2 is used.
- 7. Choose if the UMI clustering step is required, only if you have used UMIs during the library-prep.
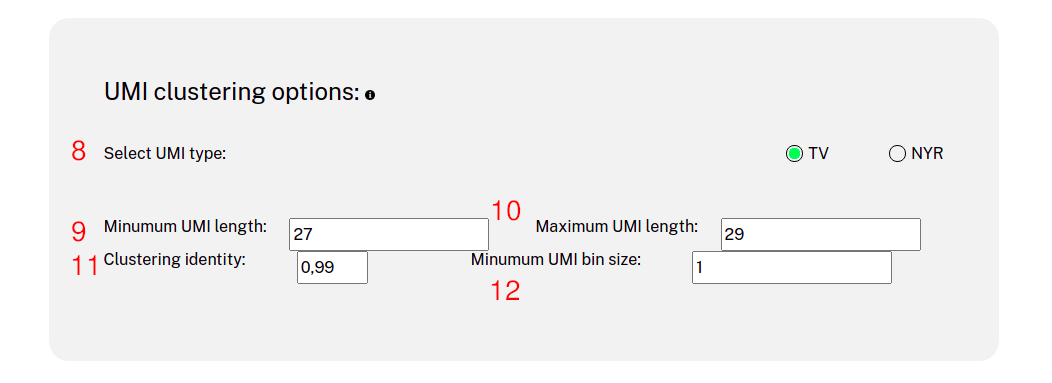
- 8. If the UMI clustering step is required, you can select the UMI type. Currently only TV (TTTVVVVTTVVVVTTVVVVTTVVVVTTT) and NYR (NNNYRNNNYRNNNYRNNN) UMIs are available.
- 9 & 10. Select or modify the maximum and minimum UMI length. By default the UMI length +/- 1 bp is used.
- 11. Modify the clustering identity value, by default 0.99.
- 12. Modify the minimum UMI bin size, by default 1, all UMIs are selected regardless of the UMI bin size.
Read structure
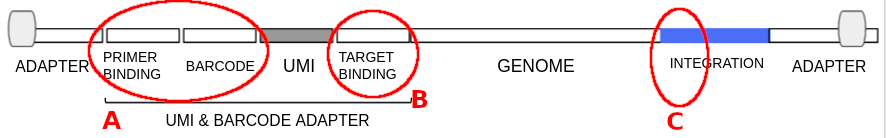
After the library-pre protocol and Oxford Nanopore sequencing, the reads will have the following structure.
Sequencing adapters are ligated at both ends.
The UMI & barcode adapter used for PCR amplifications will be presend in one end, consisting of three main parts:
- A. The primer binding site (complementary to the sequencing adapter) and the barcode sequence.
- UMI. The unimolecular identifyer sequence.
- B. The target binding site (complementary between PCR 1 and PCR2 adapters).
This adapter is bound to the genome and at the other end of the read we will find the integration site.
- C. The sequence corresponding to the paylod (or integration site) right next to the juntion is required by the analysis pipeline in the section "Ontarget primer sequence" of the input.
Results
When a run end, a compressed file will be generated for you to download the results.
Results are organized in folders.
- FILTERED. Contains quality filtered and trimmed reads.
- MAPPED. Contains SAM and BAM files for read alignment inspection. Alignments can be visualized with any genome browser program such as IGV or Geneious.
- PEAKS.
- *_peaks.bed Bed file containing sigfinicant insertions. The columns correspond to chromosome, start, end, number of reads per insertion, DHD distance to a reference peak.
- *_filtered_peakSite.bed Bed file containing significant insertions with only the predicted insertion point instead of a range.
- *_prePeakCalling_sorted_insertions.bed Bed file with all genomic regions with mapped reads, contains significant and non significant insertions or noise.
- STATS. Contains one folder per sample with histograms of raw read length or Nanopore sequencing quality.
- UMI. Containes extracted UMIs from each read and the consensus reads after UMI clustering.
- OUTPUT. Contains plots and tables of gene feature annotations and unanchored peak calling.
- *_unanchored_peaks.bed Bed file containing unanchored insertions.