CRISPR-A
documentation
CRISPR-A is a platform to assess the quality of gene editing using next generetion sequencing data. It provides an easy, sensitive, and comprehensive analysis of gene editing results. Results presition can be increased with the use of unique molecular identifiers (UMIs) and spikes or synthetic edits.
CRISPR-A: a nextflow pipeline
In addition to the web implementation, the nextflow pipeline is available at: https://bitbucket.org/synbiolab/crispr-a_nextflow/.
The analysis algorithm is composed of three mandatory steps:
1) reads pre-processing for quality assessment,
2) reads alignment against reference amplicon, and
3) edit calling.
There are other optional processes: UMI clustering, reference discovery, size bias correction, and noise subtraction based on an empirical model from negative control samples.
This is a versatile tool that can be used in a broad list of genome editing experiments without the need of specifying much information.
Analysed data
Which kind of data can you analysed with CRISPR-A?
Targeted Next Generation Sequencing data, preferentially from sequencing by synthesis like Illumina.
Which can of experiments I can analyse?
- Double strand break (DSB) with CRISPR-based technologies
- Deletions produced with double nicking strategies
- Base editing (BE)
- Prime editing (PE)
- Homology directed repair (HDR)
- Predicted off-target sites
In summary, any kind of edit small enough to be characterized through targeted sequencing.
Which is the recomended coverage?
- For clonal populations it is recommended to have at least 500 reads
- For cell bulks it is recommended to have at least 2000 reads
Even though, it will highly depend on the quality of the sample, complexity of the amplicon sequence, diversity of outcomes...
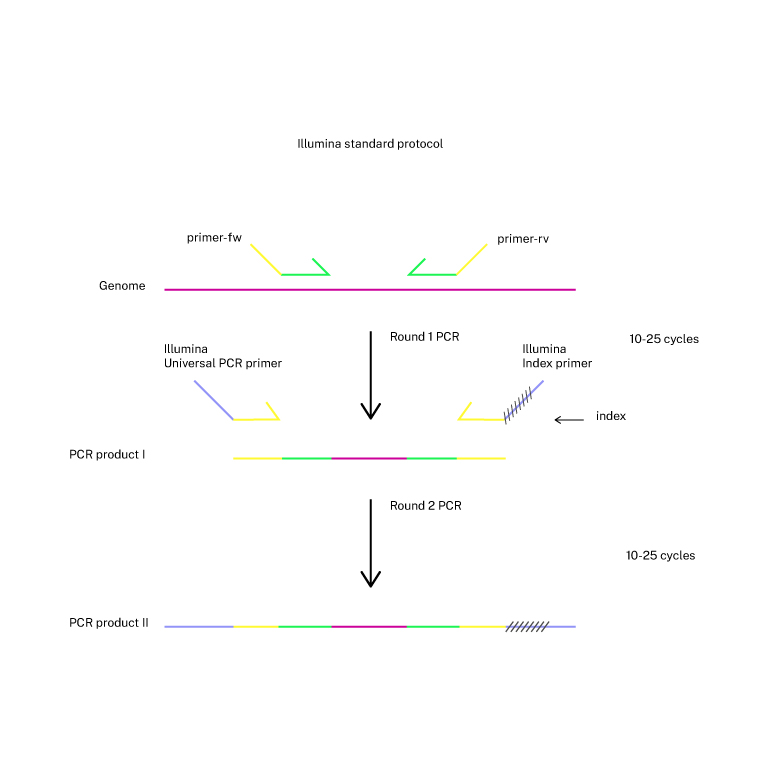
PCR amplification of the target locus
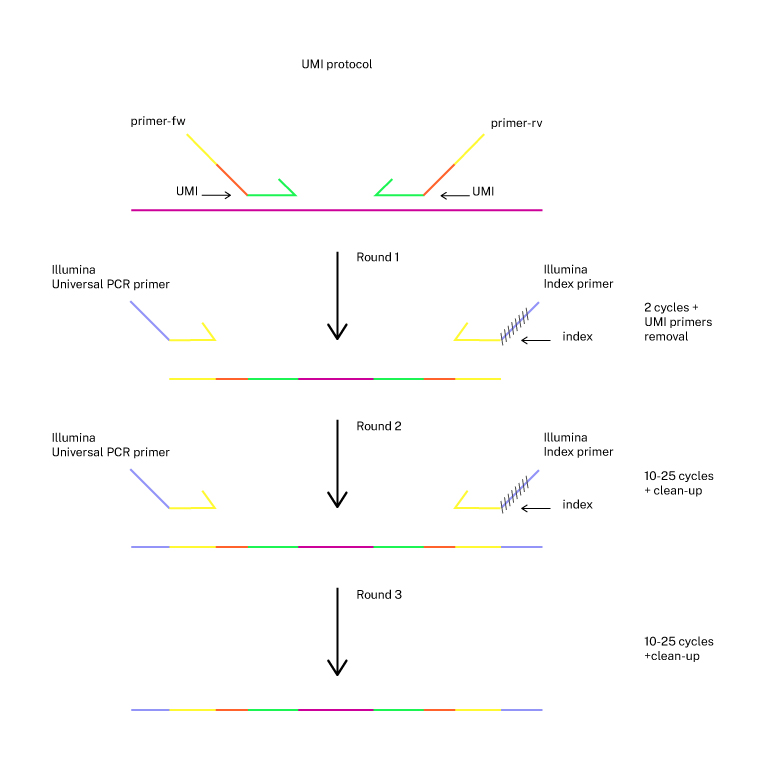
2-step PCR is carried out from a population of cells. If UMIs are included, a first 2 cycles PCR will be followed by a second and third PCR (3-step PCR). The r>
First PCR (primers with specific sequences targeting the edited locus with overhangs matching NEB universal primers):
primer-fw: 5' ACACTCTTTCCCTACACGACGCTCTTCCGATCT - (specific forward primer) 3'
primer-rv: 5' GACTGGAGTTCAGACGTGTGCTCTTCCGATCT - (specific reverse primer) 3'
First PCR for UMIs protocol (primers with specific sequences targeting the edited locus with UMIs and overhangs matching NEB universal primers):
primer-fw: 5' ACACTCTTTCCCTACACGACGCTCTTCCGATCT TTTVVVVTTVVVVTTVVVVTTVVVVTTT - (specific forward primer) 3'
primer-rv: 5' GACTGGAGTTCAGACGTGTGCTCTTCCGATCT TTTVVVVTTVVVVTTVVVVTTVVVVTTT - (specific reverse primer) 3'
Second PCR (general primers encoding barcodes, NEB P/N E7335S):
Illumina universal primer: 5' AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC*T-3'
Index N primer: 5' CAAGCAGAAGACGGCATACGAGAT NNNNNNNN GACTGGAGTTCAGACGTGTGCTCTTCCGATC-s-T-3'
Third PCR in UMIs protocol uses the primers of the second PCR.
Illumina Miseq Sequencing
Most the standard v2, and v3 Miseq kits would produce optimal results. We recommend to design the experiment following these guidelines:

Design PCR primers so that the genotyping area is localized around the center of the read

Reads of length 150bp or more will allow the detection of more complex structural variants
Paired end reads produces superior quality analysis as both reads are used for analysis
We usually use Illumina based technologies but any next generation sequencing platform providing reads with the corresponding quality (FASTQ) could be used.
CRISPR-A 3-click analysis
The CRISPR-A pipeline consists of five different steps: Miseq reads quality control, mapping, indel calling; targeted changes (HR, PE, BE...) and NHEJ estimation, and graphical representation. First, the sequencing reads are uploaded, the 3' end is trimmed of nucleotides with a Phred score lower than 20, and any reads shorter than 80bp are discarded. Second, the reads are mapped to the reference sequence provided by the user using minimap2 with optimized parameters for indel detection. If the user does not gives the amplicon referenc sequence, it can be infered from the genome of reference. If the user inputs paired end reads, the reads will be merged with PEAR. Most users will supply paired end reads, as all new Illumina kits only support paired ends. However, CRISPR-A single end compatibility will be maintained to support all possible experimental setups.
In the third step, R statistical language is used to process the mapped results, to call the insertions and deletions. Forth, CRISPR-A looks for reads matching the expected sequence, generating other variants. Fifth, R statistical language is used to produce a report that integrates plots for indels, and other expected edits. In addition, quantification precision can be increased with the use of UMIs and spikes as as a control. Noise can also be removed through and empiric model usig negative control (mock) samples.
Instead of analysing real data, a reference sequence and the cut site position can be given to simulate edition in a region of interest.
Website usage
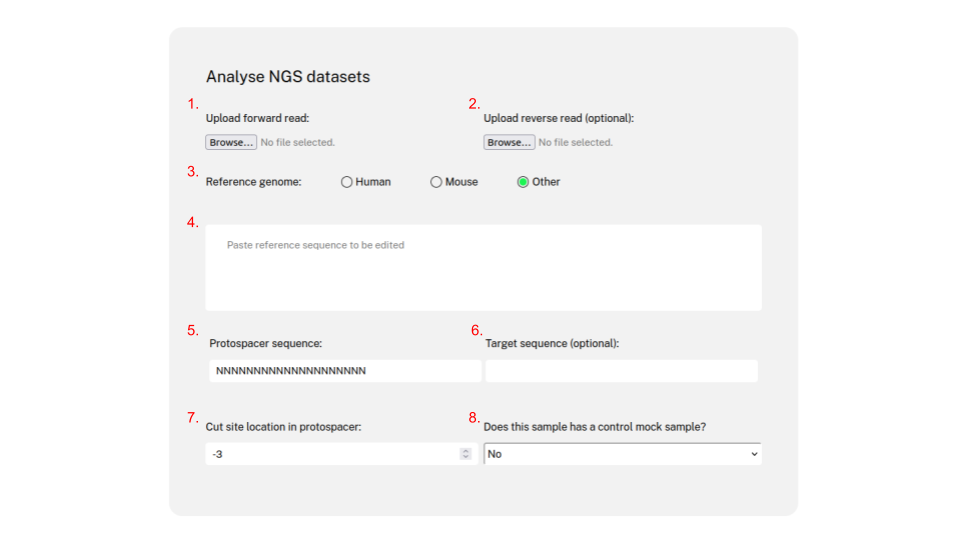
- 1. Insert forward read: Upload forward read file in fastq, or compressed fastq format.
- 2. Insert reverse read: Upload reverse read file in fastq, or compressed fastq format. This is optional. You can leave this box blank if you prefer to work with a single end library.
- 3. Indicate reference genome organisme. If the the amplified region comes from human or mouse genome, the amplicon sequence can be infered and reference sequence is not required.
- 4. Paste reference sequence: This is the sequence of the unmodified locus. We recommend you paste the full sequence of the PCR product that you are amplifying.
- 5. Paste protospacer sequence. This is optional and will be used for visualitzation.
- 6. Paste target sequence: This should be the sequence of your template DNA, donor for homologous recombination, or any expected particular modification. This is optional. You can leave this box blank if you are not interested in a certain change.
- 7. Indicate cut site location relative to the protospacer. Use "-" when counting from the end and "+" from the beggining. E.g.: For SpCas9 it is -3.
- 8. Indicate which is your negative control to remove noise, if you have used it.
- 9. Indicate if reads must be clusterized by UMIs or not to improve quantification precision and avoid PCR duplicates.
- 10. Indicate if spikes have to be used to improve quantification precision and correct quantification by edit length.
- 11. Paste reference sequence. This is optional and will just be used to simulat edits if no real data is given.
- 12. Indicate in which location of the reference sequence will take place the cut.
- 13. Paste protospacer sequence. This is optional and will be used for visualitzation.
Example
This is an example of an editing experiment carried on the AAVS human locus. We provide the reference sequence, the protospacer sequence and the fastq files containing the edited data.
Click the button to see the results report produced by CRISPR-A: